
T cellular epigenetic transforming proceeds past organismal lifespanTo resolve if epigenetic metrics of mobile growing older go beyond organismal lifespan, we hired an growing older style during which CD8+ T cells a ways exceed the standard lifetime (LT) of the host13 (Fig. 1a). The mice have been immunized with 3 heterologous top–enhance–enhance immunizations (VSVnj, VVn and VSVind; Strategies), with 30 as opposed to 60 day periods between infections. Congenically distinct VSV-specific CD8+ T cells have been then adoptively transferred into naive mice that then underwent 3 further immunizations with the heterologous virus schema. This adoptive switch and boosting cycle was once repeated as much as 16 extra instances over 10 years to determine reminiscence T cells ranging between 0.5 and ~4 mouse LTs (herein, 0.5 to 4× LT, in response to a mean lifespan of two.5 years) relative to the unique progenitor cells (Fig. 1a). The ML iteratively stimulated reminiscence CD8+ T cells care for a strong skill to proliferate all through the 51-boost regimen13. Particularly, the iteratively stimulated T cells preserved their proliferative capability (Fig. 1b). The usage of purified ML reminiscence T cells from this style we carried out complete genome bisulfite sequencing to determine a nucleotide-resolution map of the DNA methylation adjustments that happen between physiologically elderly and ML reminiscence T cells. We first assessed the epigenetic dating between younger reminiscence T cells and the ML reminiscence T cells through acting a main element research (PCA) of the highest 3,000 maximum variable CpG websites (Fig. 1c). The effects from this research printed a definite segregation between younger reminiscence CD8+ T cells as opposed to the ML reminiscence T cells, documenting CD8+ T cells’ skill to go through additional important epigenetic amendment all through and a ways exceeding organismal lifespan. Those effects additionally point out that analyses carried out with younger reminiscence T cells fail to totally seize epigenetic techniques related to the later growing older occasions (Fig. 1c). We subsequent tested the genome-wide methylation distribution throughout T cells starting from naive to 4 mouse LTs, with LT being explained because the chronologic time spanning from building of reminiscence T cells established through the primary spherical of top–enhance immunizations to the present boosted inhabitants of T cells. Particularly, an important relief of worldwide DNA methylation was once seen between ‘younger’ (~3 months put up an infection) reminiscence T cells as opposed to reminiscence T cells analyzed at complex age (~14 months this is, 0.5× LT) and a couple of (>100 months, this is, 4× LT) organismal LTs (Fig. 1d). Those findings prolong earlier observations concluding that growing older is related to a generalized lower in DNA methylation19. Additional interrogation of the DNA methylation profiles printed exaggerated DNA demethylation of effector loci and inhibitory receptors that corresponded with ATAC sequencing-defined adjustments in chromatin accessibility and transcript expression (Prolonged Knowledge Fig. 1a,d).Fig. 1: Epigenetic profiles of physiologically elderly and ML purposeful reminiscence T cells. a, Schema for producing ML reminiscence CD8+ T cells. Naive mice have been inflamed with VSV producing antigen-specific reminiscence CD8+ T cells. Heterologous VSV an infection and boosting of the reminiscence T cells was once carried out after 30 as opposed to 60 days put up earlier an infection. After 3 infections, the antigen-specific reminiscence CD8+ T cells have been transferred into congenically distinct naive mice, and the boosting technique was once repeated over a period of time that yielded reminiscence T cells with age ranging between 0.5 and ~4 mouse lifetime (LT) equivalents. For younger reminiscence, n = 6; 0.5× LT, n = 3; 2–3× LT, n = 5; and four× LT, n = 3. b, Consultant FACS research of cellular hint violet (CTV) dilution from 3° (CD45.2) and 51° (CD45.1) reminiscence T cells transferred into recipient CD45.1/2 mice. CTV label was once measured 10 days after VSV-Indiana problem of the chimeric mice. c, PCA plot of three,000 maximum variable CpG websites evaluating ML and ‘younger’ reminiscence T cells. Plot appearing main elements 1 and a pair of (PC1 and PC2). d, Violin plots appearing moderate genome-wide CpG methylation for naive, younger reminiscence and ML CD8+ T cells. The purple triangles constitute the median values. e, Heatmap appearing best ML achieve of methylation DMRs. f, GSEA the usage of cellular cycle regulators and achieve as opposed to lack of methylation DMRs for 4× LT reminiscence CD8+ T cells relative to younger reminiscence CD8+ T cells. The P price is in response to the weighted Kolmogorov–Smirnov statistic and not using a adjustment. g, Genomic and CpG island annotation of best loss and achieve of methylation ML DMRs. h, Abstract graph appearing imply methylation throughout best ML achieve of methylation DMRs relative to boosting and age of reminiscence T cells. Other lengths of resting duration additionally lead to important adjustments of imply methylation ranges. The P price is in response to the two-sided Pupil’s t-test.In spite of a generalized genome-wide relief in DNA methylation, discrete areas of the genome become closely methylated in an age-associated hierarchical way with little or no trade in chromatin accessibility (Fig. 1e and Prolonged Knowledge Fig. 1e,f). To additional interrogate epigenetic adjustments related to cumulative age and revel in in spite of retention of a T cellular’s proliferative capability, we carried out more than a few Gene Ontology (GO) and gene set enrichment analyses (GSEA). Differentially methylated areas (DMRs) between the younger and ML reminiscence T cells printed important enrichment of cellular cycle-related genes a number of the achieve of methylation DMRs (Fig. 1f). Genomic annotation of the GSEA main edges printed that achieve of methylation DMRs have been extremely enriched for promoter, UTR and exonic areas relative to the lack of methylation DMRs (Fig. 1g). A number of of those achieve of methylation occasions befell at genomic places that have been already inaccessible amongst naive T cells (Prolonged Knowledge Fig. 3b,c). Moreover, the achieve of methylation was once predominantly related to CpG islands and shores (Fig. 1g), in line with prior experiences of age-associated adjustments in CpG island methylation20,21. In line with the genomic annotation effects, we centered our consideration on characterization of the highest methylated areas a number of the iteratively boosted murine reminiscence T cells (Supplementary Desk 1). Quantification of the whole methylation for those areas in comparison to the relative LT of the samples printed a temporal dating that was once dependent at the age of cells (Fig. 1h). We subsequent sought to interrogate the impact of resting time between boosts and, thus, when put next samples that rested for 60 or 30 days after a identical selection of boosts. Particularly, T cells boosted ~50 instances with 60 day rests had a extremely important higher imply methylation amongst those DMRs as in comparison to T cells boosted ~50 instances with simplest 30 days of relaxation (Fig. 1h). On the other hand, for the reason that mouse reminiscence T cells go through a low charge of basal proliferation (continuously known as homeostatic self-renewal), this age-associated distinction might be related to cumulative proliferation. Moreover, the height growth of the boosted T cells was once persistently a lot decrease when the cells have been simplest rested for 30 days, additional indicating that ML reminiscence T cells generated from 50 boosts with 30 days relaxation periods underwent much less proliferation relative to the cells rested for 60 days between boosts (Prolonged Knowledge Fig. 2). Jointly, those effects expose that T cellular growing older ends up in the purchase of DNA methylation at cellular cycle regulator loci. Additionally, our ML style demonstrates that the epigenetic plasticity of T cells a ways exceeds organismal lifespan.Epigenetic amendment of cellular cycle regulators in elderly T cellsUpon additional exam, we famous that cyclin dependent kinase inhibitors (CDKs) Cdkn2a (p19Arf, p16Ink4a) and Cdkn2b (p15Ink4b) are a number of the genes enriched within the methylation signature (Fig. 2a and Prolonged Knowledge Fig. 3a). The deletion or silencing of this gene cluster is usually seen in more than a few cancers, particularly malignancies of lymphoid origin22,23,24,25. A abstract of the DMR methylation ranges within the Cdkn2a/b loci displays an important building up in moderate methylation ranges from the younger to ML reminiscence CD8+ T cells (Fig. 2b). On the other hand, those DMRs are got within the gene frame and exons, versus the prior to now described CpG island methylation that represses expression in settings of malignancy26. Additionally, the notable absence of promoter methylation was once accompanied through chromatin accessibility (Prolonged Knowledge Fig. 3b). Such exonic and gene frame methylation has traditionally been related to transcriptional process of those canonical senescence-associated genes24. Particularly, gene expression from those loci was once low in abundance in resting ML reminiscence T cells (Prolonged Knowledge Fig. 3c). We additionally tested the methylation standing of alternative loci coding for proteins curious about regulating replicative senescence. Certainly, demethylation of the oncogenes Mdm2, Rb1 and Cdk6 (Fig. 2c) was once inversely correlated with the achieve of methylation seen on the Cdkn2a/2b gene cluster. Not like the achieve of methylation on the Cdkn2a/2b gene cluster, the lower in DNA methylation of genes curious about tumor suppression was once related to an building up in chromatin accessibility (Prolonged Knowledge Fig. 3d). To additional interrogate the connection between T cellular proliferation and Cdkn2a gene frame methylation, we analyzed expression of the gene merchandise in Dnmt3a-knockout (KO) T cells that had skilled persistent stimulation. DNA methylation profiling showed that Dnmt3a regulates the gene frame methylation of Cdkn2a and Cdkna2b. Importantly, Cdkn2a gene expression was once upper in wildtype (Rosa inappropriate KO) T cells relative to Dnmt3a KO T cells, while Mki67 gene expression (a surrogate for proliferation) was once upper in Dnmt3a KO T cells (Prolonged Knowledge Fig. 3e,f), in line with our prior publication27.Fig. 2: DNA methylation profiles of replicative senescence-associated genes.
a, Schema for producing ML reminiscence CD8+ T cells. Naive mice have been inflamed with VSV producing antigen-specific reminiscence CD8+ T cells. Heterologous VSV an infection and boosting of the reminiscence T cells was once carried out after 30 as opposed to 60 days put up earlier an infection. After 3 infections, the antigen-specific reminiscence CD8+ T cells have been transferred into congenically distinct naive mice, and the boosting technique was once repeated over a period of time that yielded reminiscence T cells with age ranging between 0.5 and ~4 mouse lifetime (LT) equivalents. For younger reminiscence, n = 6; 0.5× LT, n = 3; 2–3× LT, n = 5; and four× LT, n = 3. b, Consultant FACS research of cellular hint violet (CTV) dilution from 3° (CD45.2) and 51° (CD45.1) reminiscence T cells transferred into recipient CD45.1/2 mice. CTV label was once measured 10 days after VSV-Indiana problem of the chimeric mice. c, PCA plot of three,000 maximum variable CpG websites evaluating ML and ‘younger’ reminiscence T cells. Plot appearing main elements 1 and a pair of (PC1 and PC2). d, Violin plots appearing moderate genome-wide CpG methylation for naive, younger reminiscence and ML CD8+ T cells. The purple triangles constitute the median values. e, Heatmap appearing best ML achieve of methylation DMRs. f, GSEA the usage of cellular cycle regulators and achieve as opposed to lack of methylation DMRs for 4× LT reminiscence CD8+ T cells relative to younger reminiscence CD8+ T cells. The P price is in response to the weighted Kolmogorov–Smirnov statistic and not using a adjustment. g, Genomic and CpG island annotation of best loss and achieve of methylation ML DMRs. h, Abstract graph appearing imply methylation throughout best ML achieve of methylation DMRs relative to boosting and age of reminiscence T cells. Other lengths of resting duration additionally lead to important adjustments of imply methylation ranges. The P price is in response to the two-sided Pupil’s t-test.In spite of a generalized genome-wide relief in DNA methylation, discrete areas of the genome become closely methylated in an age-associated hierarchical way with little or no trade in chromatin accessibility (Fig. 1e and Prolonged Knowledge Fig. 1e,f). To additional interrogate epigenetic adjustments related to cumulative age and revel in in spite of retention of a T cellular’s proliferative capability, we carried out more than a few Gene Ontology (GO) and gene set enrichment analyses (GSEA). Differentially methylated areas (DMRs) between the younger and ML reminiscence T cells printed important enrichment of cellular cycle-related genes a number of the achieve of methylation DMRs (Fig. 1f). Genomic annotation of the GSEA main edges printed that achieve of methylation DMRs have been extremely enriched for promoter, UTR and exonic areas relative to the lack of methylation DMRs (Fig. 1g). A number of of those achieve of methylation occasions befell at genomic places that have been already inaccessible amongst naive T cells (Prolonged Knowledge Fig. 3b,c). Moreover, the achieve of methylation was once predominantly related to CpG islands and shores (Fig. 1g), in line with prior experiences of age-associated adjustments in CpG island methylation20,21. In line with the genomic annotation effects, we centered our consideration on characterization of the highest methylated areas a number of the iteratively boosted murine reminiscence T cells (Supplementary Desk 1). Quantification of the whole methylation for those areas in comparison to the relative LT of the samples printed a temporal dating that was once dependent at the age of cells (Fig. 1h). We subsequent sought to interrogate the impact of resting time between boosts and, thus, when put next samples that rested for 60 or 30 days after a identical selection of boosts. Particularly, T cells boosted ~50 instances with 60 day rests had a extremely important higher imply methylation amongst those DMRs as in comparison to T cells boosted ~50 instances with simplest 30 days of relaxation (Fig. 1h). On the other hand, for the reason that mouse reminiscence T cells go through a low charge of basal proliferation (continuously known as homeostatic self-renewal), this age-associated distinction might be related to cumulative proliferation. Moreover, the height growth of the boosted T cells was once persistently a lot decrease when the cells have been simplest rested for 30 days, additional indicating that ML reminiscence T cells generated from 50 boosts with 30 days relaxation periods underwent much less proliferation relative to the cells rested for 60 days between boosts (Prolonged Knowledge Fig. 2). Jointly, those effects expose that T cellular growing older ends up in the purchase of DNA methylation at cellular cycle regulator loci. Additionally, our ML style demonstrates that the epigenetic plasticity of T cells a ways exceeds organismal lifespan.Epigenetic amendment of cellular cycle regulators in elderly T cellsUpon additional exam, we famous that cyclin dependent kinase inhibitors (CDKs) Cdkn2a (p19Arf, p16Ink4a) and Cdkn2b (p15Ink4b) are a number of the genes enriched within the methylation signature (Fig. 2a and Prolonged Knowledge Fig. 3a). The deletion or silencing of this gene cluster is usually seen in more than a few cancers, particularly malignancies of lymphoid origin22,23,24,25. A abstract of the DMR methylation ranges within the Cdkn2a/b loci displays an important building up in moderate methylation ranges from the younger to ML reminiscence CD8+ T cells (Fig. 2b). On the other hand, those DMRs are got within the gene frame and exons, versus the prior to now described CpG island methylation that represses expression in settings of malignancy26. Additionally, the notable absence of promoter methylation was once accompanied through chromatin accessibility (Prolonged Knowledge Fig. 3b). Such exonic and gene frame methylation has traditionally been related to transcriptional process of those canonical senescence-associated genes24. Particularly, gene expression from those loci was once low in abundance in resting ML reminiscence T cells (Prolonged Knowledge Fig. 3c). We additionally tested the methylation standing of alternative loci coding for proteins curious about regulating replicative senescence. Certainly, demethylation of the oncogenes Mdm2, Rb1 and Cdk6 (Fig. 2c) was once inversely correlated with the achieve of methylation seen on the Cdkn2a/2b gene cluster. Not like the achieve of methylation on the Cdkn2a/2b gene cluster, the lower in DNA methylation of genes curious about tumor suppression was once related to an building up in chromatin accessibility (Prolonged Knowledge Fig. 3d). To additional interrogate the connection between T cellular proliferation and Cdkn2a gene frame methylation, we analyzed expression of the gene merchandise in Dnmt3a-knockout (KO) T cells that had skilled persistent stimulation. DNA methylation profiling showed that Dnmt3a regulates the gene frame methylation of Cdkn2a and Cdkna2b. Importantly, Cdkn2a gene expression was once upper in wildtype (Rosa inappropriate KO) T cells relative to Dnmt3a KO T cells, while Mki67 gene expression (a surrogate for proliferation) was once upper in Dnmt3a KO T cells (Prolonged Knowledge Fig. 3e,f), in line with our prior publication27.Fig. 2: DNA methylation profiles of replicative senescence-associated genes.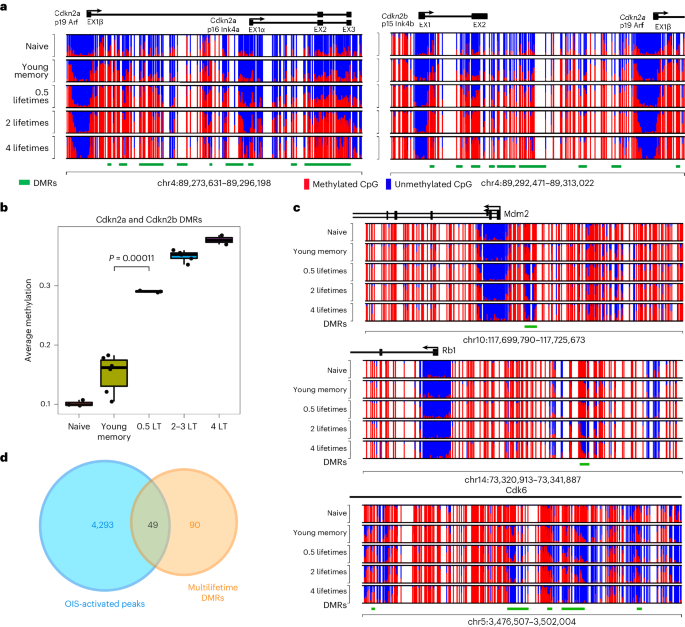 a, Consultant CpG DNA methylation plot of Cdkn2a/2b loci in naive, younger reminiscence and ML reminiscence CD8+ T cells. b, Abstract graph of overall DMR methylation a number of the Cdkn2a/b loci for younger and ML reminiscence CD8+ T cells. n = 2–5 for biologically impartial samples. The P price in response to a two-sided Pupil’s t-test. The field and hinges correspond to the primary, 2d and 0.33 quartiles, the higher whisker extends to the minimal (greatest price, higher hinge + 1.5 × interquartile vary) and the decrease whisker extends to the utmost (smallest price, decrease hinge − 1.5 × interquartile vary). c, Consultant CpG DNA methylation of Mdm2, Rb1 and Cdk6. d, Venn diagram appearing overlap between OIS-activated enhancers and best 139 DMRs.To additional assess our T cellular age epigenetic signature for genes related to replicative senescence, we subsequent cross-referenced our DMR gene record with revealed oncogene-induced senescence (OIS)-specific regulatory elements28. Of the highest achieve of methylation DMRs seen in ML T cells, over one-third (49 genes) overlap with OIS-activated enhancers, lots of which can be cellular cycle regulators (Fig. 2nd). This enrichment additional signifies that the achieve of methylation program is coupled to a mechanism that permits T cells to steer clear of replicative senescence. Different important genes of hobby recognized from the hypermethylated DMR record come with those who encode further cellular cycle regulators, comparable to Ebf3, Irx2 and Sox1 (Prolonged Knowledge Fig. 1f). Taken in combination, the epigenetic amendment of genes encoding cellular cycle regulators might keep watch over senescence of iteratively stimulated T cells and function a surrogate of T cellular proliferation.T cellular EA is uncoupled from chronologic ageGiven that the revolutionary enrichment of DNA methylation at cellular cycle regulatory genes was once correlated to reminiscence T cellular age and proliferative historical past, we subsequent sought to resolve whether or not our ML DNA methylation signature was once coupled to established epigenetic clocks. A linear regression research the usage of T cells remoted from our murine ML style displays a favorable correlation between the age of the cellular and the typical methylation of our core set of DMRs (Fig. 3a). In a similar fashion, the Horvath clock29 additionally displays a favorable correlation between the age of the murine T cells and the typical methylation (Fig. 3a). Evaluating the have compatibility of those linear regression fashions displays that the imply methylation of the ML ‘experiential age’ (EA) program is healthier correlated with the chronologic age of the mouse T cellular than the printed epigenetic clock. Particularly, there’s little overlap between the CpG websites used for established epigenetic clocks and our EA program (Prolonged Knowledge Fig. 4a). Established epigenetic clocks had been related to the PRC2 complex18,30, but we seen little or no overlap with those genomic areas explained from nonimmune cellular populations (Prolonged Knowledge Fig. 4b). On the other hand, after we when put next our ML techniques to revealed effector T cellular H3K27me3 changed genes31, we seen a placing enrichment amongst our ML techniques (Prolonged Knowledge Fig. 4c). Importantly, not like prior observations made with stem cells18, our T cell-specific growing older signature isn’t coupled to a decline in proliferation or effector possible. Those information show that epigenetic clocks aren’t unequivocally related to purposeful impairment.Fig. 3: Age estimation of mouse and human immune cells the usage of epigenetic clocks.
a, Consultant CpG DNA methylation plot of Cdkn2a/2b loci in naive, younger reminiscence and ML reminiscence CD8+ T cells. b, Abstract graph of overall DMR methylation a number of the Cdkn2a/b loci for younger and ML reminiscence CD8+ T cells. n = 2–5 for biologically impartial samples. The P price in response to a two-sided Pupil’s t-test. The field and hinges correspond to the primary, 2d and 0.33 quartiles, the higher whisker extends to the minimal (greatest price, higher hinge + 1.5 × interquartile vary) and the decrease whisker extends to the utmost (smallest price, decrease hinge − 1.5 × interquartile vary). c, Consultant CpG DNA methylation of Mdm2, Rb1 and Cdk6. d, Venn diagram appearing overlap between OIS-activated enhancers and best 139 DMRs.To additional assess our T cellular age epigenetic signature for genes related to replicative senescence, we subsequent cross-referenced our DMR gene record with revealed oncogene-induced senescence (OIS)-specific regulatory elements28. Of the highest achieve of methylation DMRs seen in ML T cells, over one-third (49 genes) overlap with OIS-activated enhancers, lots of which can be cellular cycle regulators (Fig. 2nd). This enrichment additional signifies that the achieve of methylation program is coupled to a mechanism that permits T cells to steer clear of replicative senescence. Different important genes of hobby recognized from the hypermethylated DMR record come with those who encode further cellular cycle regulators, comparable to Ebf3, Irx2 and Sox1 (Prolonged Knowledge Fig. 1f). Taken in combination, the epigenetic amendment of genes encoding cellular cycle regulators might keep watch over senescence of iteratively stimulated T cells and function a surrogate of T cellular proliferation.T cellular EA is uncoupled from chronologic ageGiven that the revolutionary enrichment of DNA methylation at cellular cycle regulatory genes was once correlated to reminiscence T cellular age and proliferative historical past, we subsequent sought to resolve whether or not our ML DNA methylation signature was once coupled to established epigenetic clocks. A linear regression research the usage of T cells remoted from our murine ML style displays a favorable correlation between the age of the cellular and the typical methylation of our core set of DMRs (Fig. 3a). In a similar fashion, the Horvath clock29 additionally displays a favorable correlation between the age of the murine T cells and the typical methylation (Fig. 3a). Evaluating the have compatibility of those linear regression fashions displays that the imply methylation of the ML ‘experiential age’ (EA) program is healthier correlated with the chronologic age of the mouse T cellular than the printed epigenetic clock. Particularly, there’s little overlap between the CpG websites used for established epigenetic clocks and our EA program (Prolonged Knowledge Fig. 4a). Established epigenetic clocks had been related to the PRC2 complex18,30, but we seen little or no overlap with those genomic areas explained from nonimmune cellular populations (Prolonged Knowledge Fig. 4b). On the other hand, after we when put next our ML techniques to revealed effector T cellular H3K27me3 changed genes31, we seen a placing enrichment amongst our ML techniques (Prolonged Knowledge Fig. 4c). Importantly, not like prior observations made with stem cells18, our T cell-specific growing older signature isn’t coupled to a decline in proliferation or effector possible. Those information show that epigenetic clocks aren’t unequivocally related to purposeful impairment.Fig. 3: Age estimation of mouse and human immune cells the usage of epigenetic clocks.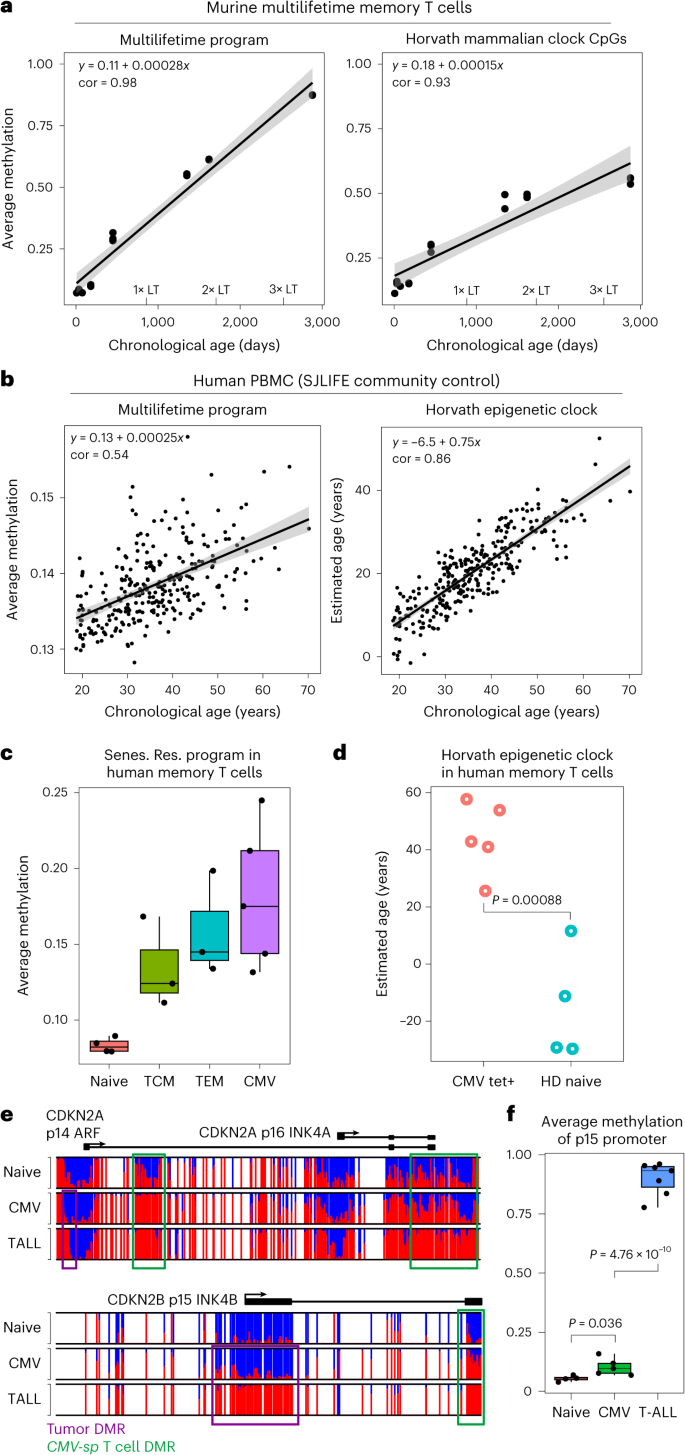 a, Left: linear regression plots of mouse T cellular age and moderate methylation ranges of ML EA-associated genes. Proper: linear regression plots of mouse T cellular age and Horvath panmammal epigenettic clock. Pearson correlation coefficient (cor) is proven in every plot. The mistake band represents the imply ± 1.96 × usual error of the imply. b, Left: linear regression plots of SJLIFE wholesome regulate (HC) chronologic age as opposed to moderate PBMC methylation stage. Proper: linear regression plots of SJLIFE HC chronologic ages and Horvath epigenetic age estimation in response to the PBMC methylation profile. Pearson cor is proven in every plot. The mistake band represents the imply ± 1.96 × usual error of the imply. c, Abstract graph of moderate ML EA-associated methylation ranges for human naive, Tcm, Tem and CMV-specific (Tetramer+) reminiscence T cellular samples. n = 3–5 for the biologically impartial samples. Senescence resistant (Senes. Res.) program approach the 139 best DMRs between endogenous and 4LT reminiscence T cells. d, Horvath epigenetic clock age estimation of CMV-specific (n = 5) and naive (n = 4) CD8+ T cells. The P price is in response to a two-sided Pupil’s t-test. The mistake bars constitute the usual deviation. e, Consultant DNA methylation plots of CDKN2A/2B gene cluster amongst naive CD8+ T cells, CMV-specific CD8+ T cells and T-ALL. f, Abstract graph for p15 promoter methylation amongst naive CD8+ T cells, CMV-specific CD8+ T cells and T-ALL. n = 4–7 for the biologically impartial samples. The P values are in response to a two-sided Pupil’s t-test. For all field plots, the field and hinges correspond to the primary, 2d and 0.33 quartiles, the higher whisker extends to the minimal (greatest price, higher hinge + 1.5 × interquartile vary) and the decrease whisker extends to the utmost (smallest price, decrease hinge − 1.5 × interquartile vary).We subsequent requested if DMRs recognized the usage of 0.5× LT cells would observe linearly with EA a number of the 2–4× LT samples. Particularly, those techniques plateaued at roughly one LT and are not able to correctly replicate EA past lifespan obstacles (Prolonged Knowledge Figs. 4d,e). Those information point out that the putative clock derived from the ML style can measure a T cellular’s proliferative historical past past organismal lifespan. To additional check the speculation that T cellular EA is self sufficient of host chronologic age, we FACS (fluorescence-activated cellular sorting) purified naive, central reminiscence (Tcm) and effector reminiscence (Tem) CD8+ T cellular subsets from 2-year-old mice (Prolonged Knowledge Figs. 4f and 8a,b) and carried out complete genome methylation research. International DMR research explained large adjustments a number of the ML as opposed to 2-year-old T cellular subsets (Supplementary Desk 2). Additional research of overlapped DMRs between ML and reminiscence T cells from 2-year-old mice, relative to naive T cells, recognized conserved adjustments related to reminiscence T cellular formation. GO research of the overlapping DMRs printed enrichment of a couple of lymphocyte activation/apoptosis-related pathways (Prolonged Knowledge Fig. 9). Explicit evaluation of the ML EA T cellular and the Horvath epigenetic clocks printed that T cellular subsets have identical ages in younger and elderly mice (Prolonged Knowledge Fig. 4g,h). We subsequent remoted 1-year-old LCMV (lymphocytic choriomeningitis virus)-specific T cells that had answered to a unmarried an infection. Very similar to the 2-year-old polyclonal reminiscence T cellular subsets, the 1-year-old antigen-specific T cells had considerably lowered epigenetic growing older signatures (Prolonged Knowledge Fig. 4g,h). Those information additional point out that epigenetic related metrics of T cellular growing older are coupled to mitotic occasions fairly than host age.We subsequent sought to resolve if the ML–EA program was once conserved in human settings of T cellular growing older. We first applied the St. Jude Existence (SJLIFE) database of wholesome individuals32 to accomplish a linear regression research between the age of the person and the imply methylation of our ML DMRs amongst overall PBMCs (peripheral blood mononuclear cells) (Fig. 3b). Certainly, there was once a favorable correlation between age of the person and imply methylation of the highest ML DMRs, in line with the sign particularly coming from the T cellular fraction of the whole PBMC. Software of the Horvath clock33 yielded a more potent correlation between chronological age and estimated age, additional confirming the facility of this revealed epigenetic clock to estimate age (Fig. 3b). On the other hand, in spite of the sure correlation, the Horvath clock looked as if it would underestimate the host age, suggesting that immune cells might age in a different way than the organism at-large. We subsequent carried out our ML–EA epigenetic clock to CD4+ and CD8+ T cellular DNA methylation information accumulated from younger and elderly people34. Particularly, we seen a better age-associated building up within the moderate methylation a number of the CD8+ T cells (Prolonged Knowledge Fig. 5a).To additional assess age estimates from the established epigenetic clock and our ML–EA program, we centered our research on particular human T cellular populations with recognized developmental states and an infection historical past. We first evaluated the ML–EA program in well-defined human CD8+ T cellular subsets from wholesome adults35,36. Particularly, Tcm and Tem CD8+ T cells have been considerably enriched for the ML–EA program relative to naive CD8+ T cells from the similar donors (Fig. 3c). To additional assess the connection between the ML–EA program and T cellular age, we remoted cytomegalovirus (CMV) particular reminiscence CD8+ T cells from 50–60-year-old folks. In line with their inflationary phenotype, those CMV-specific CD8+ T cells replicate a human environment of repeated publicity to antigen over lengthy sessions of time (Prolonged Knowledge Fig. 5b). Accordingly, the CMV-specific reminiscence CD8+ T cells had the absolute best ranges of ML–EA-associated methylation, with telomere period estimated to be similar to different reminiscence T cellular subsets. (Fig. 3c and Prolonged Knowledge Fig. 5c,g,e). In a similar fashion, the Horvath epigenetic clock estimated that the CMV-specific CD8+ T cells to have an age ranging between 20 and 50 years outdated, while the naive CD8+ T cells had age estimates round 0 years (Fig. 3d). Very similar to the murine ML CD8+ T cells, CMV-specific CD8+ T cells nonetheless retained a capability to proliferate (Prolonged Knowledge Fig. 5d) and didn’t show off indicators of malignant transformation. Given the prior affiliation between CDKN2A/2B promoter silencing with malignant transformation, we sought to match the methylation standing of the locus in CMV-specific T cells as opposed to leukemia. Whilst each T cellular populations have methylated the gene frame and exons of this locus, simplest the leukemia got methylation on the promoter area (Fig. 3e,f, Prolonged Knowledge Figs. 5f and six and Supplementary Tables 3 and four). Those information recommend that ML–EA-associated DMRs (Fig. 3e) observe T cellular proliferation without reference to host age and aren’t classical malignancy techniques.Human leukemias go through sped up epigenetic agingWe subsequent expanded our interrogation of the T cellular experiential growing older signatures through comparing it in publicly to be had DNA methylation profiles from more than a few hematological and forged tumor cohorts37,38,39,40,41. We first used the Horvath clock to determine age estimates the usage of DNA methylation profiles from sufferers with T cellular acute lymphoblastic leukemia (T-ALL), B cellular acute lymphoblastic leukemia (B-ALL), acute myeloid leukemia (AML) and melanoma (Fig. 4a,b and Prolonged Knowledge Fig. 7a). Particularly, hematologic malignancy DNA methylation profiles yielded a considerably higher age estimation than seen with melanoma. We subsequent proceeded to resolve if T-ALL subtypes have been particularly related to other age estimates. The usage of a pediatric T-ALL cohort with affected person ages ranging between 1 and 15 years outdated, we when put next the sufferers’ chronologic age and the estimated age. Moderately strikingly, a number of of the adolescent sufferers had leukemias that have been estimated to be >100 years outdated. Moreover, we famous that the malignancies with an exaggerated age prediction, together with HOXA and TLX3, have been derived from transformation occasions that befell previous in T cellular building (Fig. 4a,b). Given the present medical want for novel metrics to delineate T-ALL subtypes, we proceeded to validate the early T cellular precursor subtype the usage of DNA methylation profiles received from an impartial cohort at St Jude/ECOG (Japanese Cooperative Oncology Workforce). Once more, we seen a exceptional dichotomy in age estimates a number of the HOXA and TLX3 subtypes as opposed to the TAL1 subtypes, revealing leukemias derived from early developmental phases to have age estimates starting from ~100–200 years outdated. Thus, well-established epigenetic clocks expect T-ALL subtypes to show off hallmarks of sped up experiential growing older exceeding human lifespan.Fig. 4: T-ALL subset epigenetic age estimation.
a, Left: linear regression plots of mouse T cellular age and moderate methylation ranges of ML EA-associated genes. Proper: linear regression plots of mouse T cellular age and Horvath panmammal epigenettic clock. Pearson correlation coefficient (cor) is proven in every plot. The mistake band represents the imply ± 1.96 × usual error of the imply. b, Left: linear regression plots of SJLIFE wholesome regulate (HC) chronologic age as opposed to moderate PBMC methylation stage. Proper: linear regression plots of SJLIFE HC chronologic ages and Horvath epigenetic age estimation in response to the PBMC methylation profile. Pearson cor is proven in every plot. The mistake band represents the imply ± 1.96 × usual error of the imply. c, Abstract graph of moderate ML EA-associated methylation ranges for human naive, Tcm, Tem and CMV-specific (Tetramer+) reminiscence T cellular samples. n = 3–5 for the biologically impartial samples. Senescence resistant (Senes. Res.) program approach the 139 best DMRs between endogenous and 4LT reminiscence T cells. d, Horvath epigenetic clock age estimation of CMV-specific (n = 5) and naive (n = 4) CD8+ T cells. The P price is in response to a two-sided Pupil’s t-test. The mistake bars constitute the usual deviation. e, Consultant DNA methylation plots of CDKN2A/2B gene cluster amongst naive CD8+ T cells, CMV-specific CD8+ T cells and T-ALL. f, Abstract graph for p15 promoter methylation amongst naive CD8+ T cells, CMV-specific CD8+ T cells and T-ALL. n = 4–7 for the biologically impartial samples. The P values are in response to a two-sided Pupil’s t-test. For all field plots, the field and hinges correspond to the primary, 2d and 0.33 quartiles, the higher whisker extends to the minimal (greatest price, higher hinge + 1.5 × interquartile vary) and the decrease whisker extends to the utmost (smallest price, decrease hinge − 1.5 × interquartile vary).We subsequent requested if DMRs recognized the usage of 0.5× LT cells would observe linearly with EA a number of the 2–4× LT samples. Particularly, those techniques plateaued at roughly one LT and are not able to correctly replicate EA past lifespan obstacles (Prolonged Knowledge Figs. 4d,e). Those information point out that the putative clock derived from the ML style can measure a T cellular’s proliferative historical past past organismal lifespan. To additional check the speculation that T cellular EA is self sufficient of host chronologic age, we FACS (fluorescence-activated cellular sorting) purified naive, central reminiscence (Tcm) and effector reminiscence (Tem) CD8+ T cellular subsets from 2-year-old mice (Prolonged Knowledge Figs. 4f and 8a,b) and carried out complete genome methylation research. International DMR research explained large adjustments a number of the ML as opposed to 2-year-old T cellular subsets (Supplementary Desk 2). Additional research of overlapped DMRs between ML and reminiscence T cells from 2-year-old mice, relative to naive T cells, recognized conserved adjustments related to reminiscence T cellular formation. GO research of the overlapping DMRs printed enrichment of a couple of lymphocyte activation/apoptosis-related pathways (Prolonged Knowledge Fig. 9). Explicit evaluation of the ML EA T cellular and the Horvath epigenetic clocks printed that T cellular subsets have identical ages in younger and elderly mice (Prolonged Knowledge Fig. 4g,h). We subsequent remoted 1-year-old LCMV (lymphocytic choriomeningitis virus)-specific T cells that had answered to a unmarried an infection. Very similar to the 2-year-old polyclonal reminiscence T cellular subsets, the 1-year-old antigen-specific T cells had considerably lowered epigenetic growing older signatures (Prolonged Knowledge Fig. 4g,h). Those information additional point out that epigenetic related metrics of T cellular growing older are coupled to mitotic occasions fairly than host age.We subsequent sought to resolve if the ML–EA program was once conserved in human settings of T cellular growing older. We first applied the St. Jude Existence (SJLIFE) database of wholesome individuals32 to accomplish a linear regression research between the age of the person and the imply methylation of our ML DMRs amongst overall PBMCs (peripheral blood mononuclear cells) (Fig. 3b). Certainly, there was once a favorable correlation between age of the person and imply methylation of the highest ML DMRs, in line with the sign particularly coming from the T cellular fraction of the whole PBMC. Software of the Horvath clock33 yielded a more potent correlation between chronological age and estimated age, additional confirming the facility of this revealed epigenetic clock to estimate age (Fig. 3b). On the other hand, in spite of the sure correlation, the Horvath clock looked as if it would underestimate the host age, suggesting that immune cells might age in a different way than the organism at-large. We subsequent carried out our ML–EA epigenetic clock to CD4+ and CD8+ T cellular DNA methylation information accumulated from younger and elderly people34. Particularly, we seen a better age-associated building up within the moderate methylation a number of the CD8+ T cells (Prolonged Knowledge Fig. 5a).To additional assess age estimates from the established epigenetic clock and our ML–EA program, we centered our research on particular human T cellular populations with recognized developmental states and an infection historical past. We first evaluated the ML–EA program in well-defined human CD8+ T cellular subsets from wholesome adults35,36. Particularly, Tcm and Tem CD8+ T cells have been considerably enriched for the ML–EA program relative to naive CD8+ T cells from the similar donors (Fig. 3c). To additional assess the connection between the ML–EA program and T cellular age, we remoted cytomegalovirus (CMV) particular reminiscence CD8+ T cells from 50–60-year-old folks. In line with their inflationary phenotype, those CMV-specific CD8+ T cells replicate a human environment of repeated publicity to antigen over lengthy sessions of time (Prolonged Knowledge Fig. 5b). Accordingly, the CMV-specific reminiscence CD8+ T cells had the absolute best ranges of ML–EA-associated methylation, with telomere period estimated to be similar to different reminiscence T cellular subsets. (Fig. 3c and Prolonged Knowledge Fig. 5c,g,e). In a similar fashion, the Horvath epigenetic clock estimated that the CMV-specific CD8+ T cells to have an age ranging between 20 and 50 years outdated, while the naive CD8+ T cells had age estimates round 0 years (Fig. 3d). Very similar to the murine ML CD8+ T cells, CMV-specific CD8+ T cells nonetheless retained a capability to proliferate (Prolonged Knowledge Fig. 5d) and didn’t show off indicators of malignant transformation. Given the prior affiliation between CDKN2A/2B promoter silencing with malignant transformation, we sought to match the methylation standing of the locus in CMV-specific T cells as opposed to leukemia. Whilst each T cellular populations have methylated the gene frame and exons of this locus, simplest the leukemia got methylation on the promoter area (Fig. 3e,f, Prolonged Knowledge Figs. 5f and six and Supplementary Tables 3 and four). Those information recommend that ML–EA-associated DMRs (Fig. 3e) observe T cellular proliferation without reference to host age and aren’t classical malignancy techniques.Human leukemias go through sped up epigenetic agingWe subsequent expanded our interrogation of the T cellular experiential growing older signatures through comparing it in publicly to be had DNA methylation profiles from more than a few hematological and forged tumor cohorts37,38,39,40,41. We first used the Horvath clock to determine age estimates the usage of DNA methylation profiles from sufferers with T cellular acute lymphoblastic leukemia (T-ALL), B cellular acute lymphoblastic leukemia (B-ALL), acute myeloid leukemia (AML) and melanoma (Fig. 4a,b and Prolonged Knowledge Fig. 7a). Particularly, hematologic malignancy DNA methylation profiles yielded a considerably higher age estimation than seen with melanoma. We subsequent proceeded to resolve if T-ALL subtypes have been particularly related to other age estimates. The usage of a pediatric T-ALL cohort with affected person ages ranging between 1 and 15 years outdated, we when put next the sufferers’ chronologic age and the estimated age. Moderately strikingly, a number of of the adolescent sufferers had leukemias that have been estimated to be >100 years outdated. Moreover, we famous that the malignancies with an exaggerated age prediction, together with HOXA and TLX3, have been derived from transformation occasions that befell previous in T cellular building (Fig. 4a,b). Given the present medical want for novel metrics to delineate T-ALL subtypes, we proceeded to validate the early T cellular precursor subtype the usage of DNA methylation profiles received from an impartial cohort at St Jude/ECOG (Japanese Cooperative Oncology Workforce). Once more, we seen a exceptional dichotomy in age estimates a number of the HOXA and TLX3 subtypes as opposed to the TAL1 subtypes, revealing leukemias derived from early developmental phases to have age estimates starting from ~100–200 years outdated. Thus, well-established epigenetic clocks expect T-ALL subtypes to show off hallmarks of sped up experiential growing older exceeding human lifespan.Fig. 4: T-ALL subset epigenetic age estimation.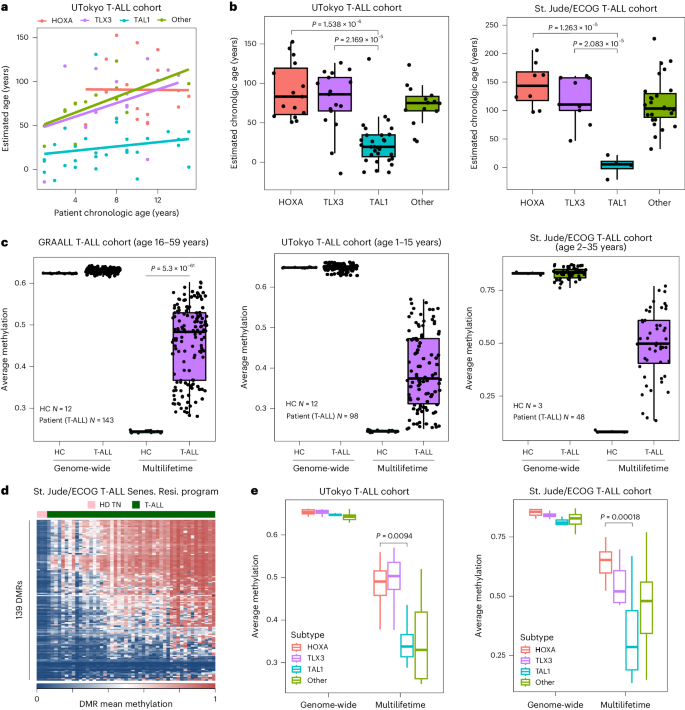 a,b, Abstract graphs of affected person chronologic age (a) as opposed to estimated tumor age (b) in response to T-ALL subset the usage of the Horvath epigenetic clock (n = 13–31 within the UTokyo cohort and n = 4–28 within the St. Jude cohort). The P values are in response to a two-sided Pupil’s t-test. c, Abstract field plots for the T-ALL affected person tumor genome-wide DNA methylation ranges as opposed to EA techniques amongst 3 impartial cohorts (GRAALL n = 143, St. Jude/ECOG n = 48 and UTokyo n = 98). The P price is in response to a two-sided Pupil’s t-test. d, DNA methylation stage heatmap of ML EA techniques for St. Jude/ECOG T-ALL sufferers. e, Abstract graph of ML EA techniques amongst established T-ALL affected person subtypes. The P values are in response to two-sided Pupil’s t-tests evaluating HOXA and TAL1 subtypes (n = 13–31 within the UTokyo cohort and n = 4–28 within the St. Jude cohort. For all field plots, the field and hinges correspond to the primary, 2d and 0.33 quartiles, the higher whisker extends to the minimal (greatest price, higher hinge + 1.5 × interquartile vary) and the decrease whisker extends to the utmost (smallest price, decrease hinge − 1.5 × interquartile vary).We subsequent interrogated the more than a few most cancers cohorts the usage of our murine ML–EA methylation signature (Fig. 4c and Prolonged Knowledge Fig. 7b,c). Comparability of the genome-wide moderate stage of methylation between wholesome controls as opposed to the malignancy cohorts printed that the ML–EA program was once considerably enriched within the sufferers with hematological malignancies (Fig. 4c and Prolonged Knowledge Fig. 7b,c). Particularly, T-ALL sufferers had the best enrichment of the ML–EA program relative to the B-ALL and AML-derived samples (Fig. 4c and Prolonged Knowledge Fig. 7b,c). The powerful enrichment of the ML–EA program amongst lymphocytic leukemias additional paperwork a hyperlink between a T cells’ putative proliferative historical past and epigenetic repression of cellular cycle regulators.Whilst there was once certainly important enrichment of the ML–EA program amongst all 3 impartial T-ALL cohorts, every cohort possessed a placing level of heterogeneity within the relative stage of the signature (Fig. 4c,d). To additional interrogate this heterogeneity, we analyzed the ML–EA program in response to the recognized T-ALL subtypes. Very similar to the age estimate, our ML–EA program was once maximum enriched amongst T-ALL derived early in T cellular building, particularly leukemias with HOXA and TLX3 genetic mutations (Fig. 4e). Those information jointly record that the ML–EA methylation techniques can delineate human T-ALL subtypes in response to T cellular maturation degree and that the buildup of epigenetic growing older metrics is uncoupled from replicative senescence.
a,b, Abstract graphs of affected person chronologic age (a) as opposed to estimated tumor age (b) in response to T-ALL subset the usage of the Horvath epigenetic clock (n = 13–31 within the UTokyo cohort and n = 4–28 within the St. Jude cohort). The P values are in response to a two-sided Pupil’s t-test. c, Abstract field plots for the T-ALL affected person tumor genome-wide DNA methylation ranges as opposed to EA techniques amongst 3 impartial cohorts (GRAALL n = 143, St. Jude/ECOG n = 48 and UTokyo n = 98). The P price is in response to a two-sided Pupil’s t-test. d, DNA methylation stage heatmap of ML EA techniques for St. Jude/ECOG T-ALL sufferers. e, Abstract graph of ML EA techniques amongst established T-ALL affected person subtypes. The P values are in response to two-sided Pupil’s t-tests evaluating HOXA and TAL1 subtypes (n = 13–31 within the UTokyo cohort and n = 4–28 within the St. Jude cohort. For all field plots, the field and hinges correspond to the primary, 2d and 0.33 quartiles, the higher whisker extends to the minimal (greatest price, higher hinge + 1.5 × interquartile vary) and the decrease whisker extends to the utmost (smallest price, decrease hinge − 1.5 × interquartile vary).We subsequent interrogated the more than a few most cancers cohorts the usage of our murine ML–EA methylation signature (Fig. 4c and Prolonged Knowledge Fig. 7b,c). Comparability of the genome-wide moderate stage of methylation between wholesome controls as opposed to the malignancy cohorts printed that the ML–EA program was once considerably enriched within the sufferers with hematological malignancies (Fig. 4c and Prolonged Knowledge Fig. 7b,c). Particularly, T-ALL sufferers had the best enrichment of the ML–EA program relative to the B-ALL and AML-derived samples (Fig. 4c and Prolonged Knowledge Fig. 7b,c). The powerful enrichment of the ML–EA program amongst lymphocytic leukemias additional paperwork a hyperlink between a T cells’ putative proliferative historical past and epigenetic repression of cellular cycle regulators.Whilst there was once certainly important enrichment of the ML–EA program amongst all 3 impartial T-ALL cohorts, every cohort possessed a placing level of heterogeneity within the relative stage of the signature (Fig. 4c,d). To additional interrogate this heterogeneity, we analyzed the ML–EA program in response to the recognized T-ALL subtypes. Very similar to the age estimate, our ML–EA program was once maximum enriched amongst T-ALL derived early in T cellular building, particularly leukemias with HOXA and TLX3 genetic mutations (Fig. 4e). Those information jointly record that the ML–EA methylation techniques can delineate human T-ALL subtypes in response to T cellular maturation degree and that the buildup of epigenetic growing older metrics is uncoupled from replicative senescence.













