

Demographic abstract of the retrieved genomes of the SARS-CoV-2To examine the spectrum of nucleotide (NT) and amino acid (AA) mutations and their results in other variants of the SARS-CoV-2, sequenced from COVID-19 deceased sufferers, we retrieved 243,270 entire genome series (WGS) with prime learn protection (> 29,000 bp) from the worldwide initiative on sharing all influenza knowledge (GISAID) as much as February 2023. After a thorough filtering of those genomes, 5724 entire genomes belonged to COVID-19 deceased sufferers from other demographics had been decided on for additional research. Those WGS knowledge (n = 5724) comprised SARS-CoV-2 genome sequences from 123 international locations and 5 continents (e.g., Asia, Africa, Europe, North The united states and South The united states) of the globe. The geographical distribution of the SARS-CoV-2 WGS from deceased COVID-19 other folks finds that 33.4% of the genomes had been sequenced from North The united states adopted via 28.8% from Europe, 19.9% from South The united states, 17.7% from Africa, and zero.7% from Asia (Fig. 1A).Determine 1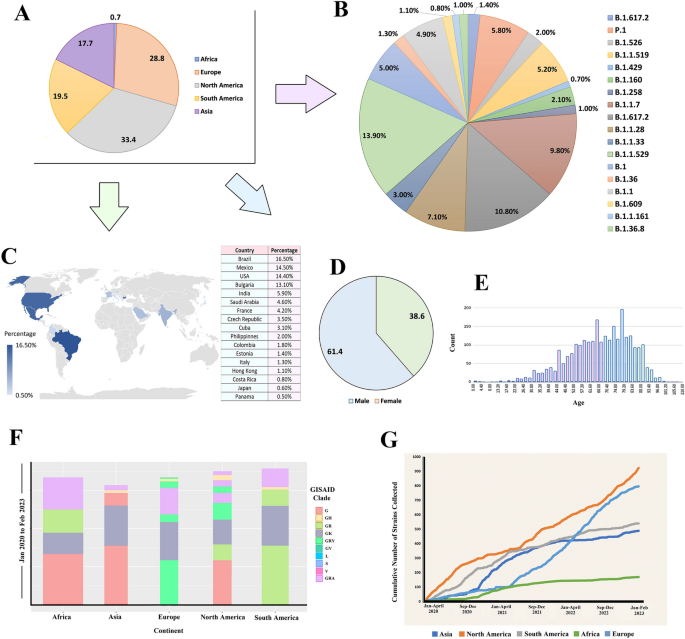 Retrieved SARS-COV-2 entire genome sequences (WGS) received from deceased COVID=19 sufferers international from the worldwide initiative on sharing all influenza database (GISAID). (A) SARS-CoV-2 genomes submitted to the GISAID from 5 continents of the globe between January 2020 and February 2023. (B) Distribution of sequences all over a number of lineages right through the time span. (C) SARS-CoV-2 genomes sequenced via other international locations from deceased sufferers. (D) Gender-wise (female and male) and (E) age-wise distribution of the chosen sequences. (F) Distribution of various clades in 5 continents. (G) Cumulative selection of lines retrieved right through the period of time from 5 continents.We discovered B.1.1.529 (Omicron) as essentially the most prevalent (13.90%) amongst all of the lineages detected while B.1.617.2 (Delta) and B.1.1.7 (Alpha) had been additionally considerably prevalent; comprising 10.80% and 9.80% of the sequences, respectively (Fig. 1B). On the other hand, B.1.1.28 (Brazilian variant), P.1 (Gamma), and B.1.1.519 (Mexican variant) had been additionally seen to be essential in 7.10, 5.80 and 5.00% of the SARS-CoV-2 genomes, respectively (Fig. 1B). Via evaluating those knowledge in keeping with other international locations of beginning, we discovered that Brazil contributed the best (16.5%) quantity of SARS-CoV-2 WGS knowledge sequenced from deceased COVID-19 sufferers, adopted via Mexico (14.5%), america (14.4%), Bulgaria (13.1%), and India (5.1%). The bottom quantity of WGS (0.5%) used to be sequenced from deceased COVID-19 sufferers in Panama (Fig. 1C). In keeping with the metadata, 61.4% of deceased COVID-19 sufferers had been male whilst 38.6% had been feminine (Fig. 1D) with a mean age of roughly 70 years (Fig. 1E), which used to be now not explicitly said within the GISAID. Additional research demonstrated that male sufferers with a mean age of 55 years had upper COVID-19 possibility than feminine sufferers.The “G” clade of the SARS-CoV-2 used to be discovered to be predominated within the deceased COVID-19 sufferers of the Asian, African and North American areas whilst lots of the dying circumstances in Europe had been registered with “GRY” clade. Against this, maximum dying circumstances had been registered for “GR” clade within the South American continent (Fig. 1F). Via having a look on the cumulative dying circumstances registered all over find out about duration (from January 2020 to February 2023), we discovered that lots of the WGS knowledge (roughly 1000 sequences) from deceased COVID-19 sufferers had been sequenced from the North American citizens. Even though all of the international locations had submitted their WGS regardless of regional barrier, African areas are discovered because the lowest conceivable knowledge producing zone from deceased COVID-19 sufferers (lower than 200 sequences) (Fig. 1G). Related demographic and clinical knowledge are described in Information S1.Phylogenetic variety of the SARS-CoV-2 genomes of the deceased COVID-19 patientsTo decide phylogenetic traits of the SARS-CoV-2 genomes of the deceased COVID-19 sufferers, we constructed a most chance (ML) tree in accordance with aligned complete duration sequences the usage of Nextclade Internet 2.14.1 web-based software ( (Fig. 2, Information S2). The WGS knowledge assembled from deceased COVID-19 sufferers world wide confirmed the formation of 21 Nextstrain clades, together with 4 VOCs: 20I (alpha, V1), 20H (Beta, V2), 20J (Gamma, V3), and 21A, 21I, and 21J (Delta V4). As well as, the find out about genomes belonged to the VOIs, comparable to 21C (Epsilon), 21G (Lambda), and 21H (Mu), different assigned and unassigned clades together with 21B (Kappa), 21F (Lota), 20E (EU1), 19A, 19B, 20A, 20B, 20C, 20D, and 20G (Fig. 2A). The viral clade distribution of the find out about genomes represented one of the most SARS-CoV-2 genetic clades that had been circulating international right through January 2020 to February 2023. We additional explored the range of SARS-CoV-2 genomes via evaluating the gap matrix of the SARS-CoV-2 lines of the deceased sufferers to the Wuhan-Hu-1/NC 045512 reference pressure. Nextstrain classification published that clade 20I (Alpha, V1) used to be prevalent in 20% of the find out about genomes, while clade 20H (Beta, V2) and 20J (Gamma, V3) had been discovered to be prevalent in 2.42% and six.33% genomes, respectively (Fig. 2B). Additionally, clade 21A, 21I, and 21J of the Delta variant (B.1.617.2) accounted for 1.48% of the find out about genomes whilst clade 21F of the Lota variant used to be represented via 1.45% genomes. On the other hand, handiest 4 SARS-CoV-2 genomes in our investigation belonged to the Kappa variant (B.1.617.1), which used to be prevalent in India and possessed 3 vital alterations on the websites of L452R, E484Q, and P681R. As well as, 25, 3, and 16 sequences had been belonged to Epsilon (21C), Lambda (21G), and Mu (21H), respectively, that are categorised as “VOI.” Probably the most prevalent clades in our research had been 20B (30%) adopted via 20A (22.0%). On this research, essentially the most mutational frequency used to be seen within the spike protein area adopted via ORF1a fragment. While the least adjustments had been seen in ORF1b area. The best height used to be seen within the N portion (Fig. 2B).Determine 2
Retrieved SARS-COV-2 entire genome sequences (WGS) received from deceased COVID=19 sufferers international from the worldwide initiative on sharing all influenza database (GISAID). (A) SARS-CoV-2 genomes submitted to the GISAID from 5 continents of the globe between January 2020 and February 2023. (B) Distribution of sequences all over a number of lineages right through the time span. (C) SARS-CoV-2 genomes sequenced via other international locations from deceased sufferers. (D) Gender-wise (female and male) and (E) age-wise distribution of the chosen sequences. (F) Distribution of various clades in 5 continents. (G) Cumulative selection of lines retrieved right through the period of time from 5 continents.We discovered B.1.1.529 (Omicron) as essentially the most prevalent (13.90%) amongst all of the lineages detected while B.1.617.2 (Delta) and B.1.1.7 (Alpha) had been additionally considerably prevalent; comprising 10.80% and 9.80% of the sequences, respectively (Fig. 1B). On the other hand, B.1.1.28 (Brazilian variant), P.1 (Gamma), and B.1.1.519 (Mexican variant) had been additionally seen to be essential in 7.10, 5.80 and 5.00% of the SARS-CoV-2 genomes, respectively (Fig. 1B). Via evaluating those knowledge in keeping with other international locations of beginning, we discovered that Brazil contributed the best (16.5%) quantity of SARS-CoV-2 WGS knowledge sequenced from deceased COVID-19 sufferers, adopted via Mexico (14.5%), america (14.4%), Bulgaria (13.1%), and India (5.1%). The bottom quantity of WGS (0.5%) used to be sequenced from deceased COVID-19 sufferers in Panama (Fig. 1C). In keeping with the metadata, 61.4% of deceased COVID-19 sufferers had been male whilst 38.6% had been feminine (Fig. 1D) with a mean age of roughly 70 years (Fig. 1E), which used to be now not explicitly said within the GISAID. Additional research demonstrated that male sufferers with a mean age of 55 years had upper COVID-19 possibility than feminine sufferers.The “G” clade of the SARS-CoV-2 used to be discovered to be predominated within the deceased COVID-19 sufferers of the Asian, African and North American areas whilst lots of the dying circumstances in Europe had been registered with “GRY” clade. Against this, maximum dying circumstances had been registered for “GR” clade within the South American continent (Fig. 1F). Via having a look on the cumulative dying circumstances registered all over find out about duration (from January 2020 to February 2023), we discovered that lots of the WGS knowledge (roughly 1000 sequences) from deceased COVID-19 sufferers had been sequenced from the North American citizens. Even though all of the international locations had submitted their WGS regardless of regional barrier, African areas are discovered because the lowest conceivable knowledge producing zone from deceased COVID-19 sufferers (lower than 200 sequences) (Fig. 1G). Related demographic and clinical knowledge are described in Information S1.Phylogenetic variety of the SARS-CoV-2 genomes of the deceased COVID-19 patientsTo decide phylogenetic traits of the SARS-CoV-2 genomes of the deceased COVID-19 sufferers, we constructed a most chance (ML) tree in accordance with aligned complete duration sequences the usage of Nextclade Internet 2.14.1 web-based software ( (Fig. 2, Information S2). The WGS knowledge assembled from deceased COVID-19 sufferers world wide confirmed the formation of 21 Nextstrain clades, together with 4 VOCs: 20I (alpha, V1), 20H (Beta, V2), 20J (Gamma, V3), and 21A, 21I, and 21J (Delta V4). As well as, the find out about genomes belonged to the VOIs, comparable to 21C (Epsilon), 21G (Lambda), and 21H (Mu), different assigned and unassigned clades together with 21B (Kappa), 21F (Lota), 20E (EU1), 19A, 19B, 20A, 20B, 20C, 20D, and 20G (Fig. 2A). The viral clade distribution of the find out about genomes represented one of the most SARS-CoV-2 genetic clades that had been circulating international right through January 2020 to February 2023. We additional explored the range of SARS-CoV-2 genomes via evaluating the gap matrix of the SARS-CoV-2 lines of the deceased sufferers to the Wuhan-Hu-1/NC 045512 reference pressure. Nextstrain classification published that clade 20I (Alpha, V1) used to be prevalent in 20% of the find out about genomes, while clade 20H (Beta, V2) and 20J (Gamma, V3) had been discovered to be prevalent in 2.42% and six.33% genomes, respectively (Fig. 2B). Additionally, clade 21A, 21I, and 21J of the Delta variant (B.1.617.2) accounted for 1.48% of the find out about genomes whilst clade 21F of the Lota variant used to be represented via 1.45% genomes. On the other hand, handiest 4 SARS-CoV-2 genomes in our investigation belonged to the Kappa variant (B.1.617.1), which used to be prevalent in India and possessed 3 vital alterations on the websites of L452R, E484Q, and P681R. As well as, 25, 3, and 16 sequences had been belonged to Epsilon (21C), Lambda (21G), and Mu (21H), respectively, that are categorised as “VOI.” Probably the most prevalent clades in our research had been 20B (30%) adopted via 20A (22.0%). On this research, essentially the most mutational frequency used to be seen within the spike protein area adopted via ORF1a fragment. While the least adjustments had been seen in ORF1b area. The best height used to be seen within the N portion (Fig. 2B).Determine 2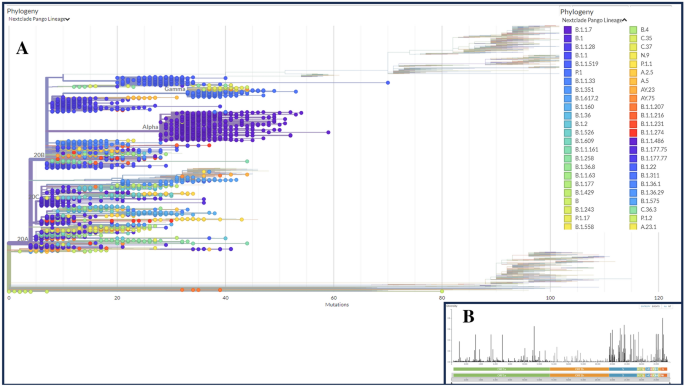 Phylogenetic analyses of the 5,724 SARS-CoV-2 genomes sequenced from the COVID-19 deceased sufferers international. (A) An in depth phylogenetic tree presenting all of the vital clades related to deceased COVID-19 sufferers. (B) Worth of the entropy trade (distribution of mutational frequency general the SARS-CoV-2 genome) all over the SARS-CoV-2 genome in accordance with mutation depend for every place. The utmost-likelihood tree used to be generated the usage of Nextclade Internet 2.14.1 web-based software ( accessed on October 10, 2023).Frequency and distribution of nucleotide mutations in SARS-CoV-2 genomes of the deceased COVID-19 patientsTo decide the frequency and distribution of nucleotide (NT) mutations, we additional analyzed 5,724 SARS-CoV-2 genomes from deceased COVID-19 sufferers of numerous demographics. We detected a mean of 12.9 NT mutations according to genome. The whole NT mutation frequencies within the SARS-CoV-2 genomes sequenced from deceased COVID-19 sufferers are proven in Fig. 3. Our complete mutational research known 35,799 NT mutations throughout all the dataset of 5724 SARS-CoV-2 genomes, of which 11,402 (best) NT mutations had been only discovered within the S gene. Conversely, the E gene possessed the bottom quantity (n = 94) of NT mutations (Fig. 3A). As well as, the selection of NT mutations in ORF1a, N, ORF1b, ORF8, ORF3a, ORF9b, ORF7a, M, ORF6, ORF7b, and E segments had been 7964, 6008, 5064, 2431, 1735, 410, 325, 177, 138, 75 and 70, respectively (Fig. 3A). The best selection of NT mutations had been known in D614G positions, adopted via N501Y, P681H, T716I, and A570D within the spike (S) protein (Fig. 3B). Of the known NT mutations, the 4 maximum widespread NT mutations comparable to T1001I, A170BD, I2230T, and T265I had been known within the N gene. Herein this find out about, we noticed 3 maximum NT mutations (e.g., P314L, E1264D, and P218L) within the ORF1b gene. As well as, E, M, ORF3a, ORF6a, ORF7b, ORF8 and ORF9b genes possessed the best selection of NT mutations at P71L, I82T, Q57H, I33T, T4OI, Y73C and Q77E positions, respectively. Different genes with vital NT mutation frequencies at particular websites integrated P71L (E-gene), T4OI (0RF7b), I33T (ORF6a), I82T (M), T40I (ORF7b), Q77E (ORF9b), Q57H (ORF3a) and Y73C (ORF8) (Fig. 3B).Determine 3
Phylogenetic analyses of the 5,724 SARS-CoV-2 genomes sequenced from the COVID-19 deceased sufferers international. (A) An in depth phylogenetic tree presenting all of the vital clades related to deceased COVID-19 sufferers. (B) Worth of the entropy trade (distribution of mutational frequency general the SARS-CoV-2 genome) all over the SARS-CoV-2 genome in accordance with mutation depend for every place. The utmost-likelihood tree used to be generated the usage of Nextclade Internet 2.14.1 web-based software ( accessed on October 10, 2023).Frequency and distribution of nucleotide mutations in SARS-CoV-2 genomes of the deceased COVID-19 patientsTo decide the frequency and distribution of nucleotide (NT) mutations, we additional analyzed 5,724 SARS-CoV-2 genomes from deceased COVID-19 sufferers of numerous demographics. We detected a mean of 12.9 NT mutations according to genome. The whole NT mutation frequencies within the SARS-CoV-2 genomes sequenced from deceased COVID-19 sufferers are proven in Fig. 3. Our complete mutational research known 35,799 NT mutations throughout all the dataset of 5724 SARS-CoV-2 genomes, of which 11,402 (best) NT mutations had been only discovered within the S gene. Conversely, the E gene possessed the bottom quantity (n = 94) of NT mutations (Fig. 3A). As well as, the selection of NT mutations in ORF1a, N, ORF1b, ORF8, ORF3a, ORF9b, ORF7a, M, ORF6, ORF7b, and E segments had been 7964, 6008, 5064, 2431, 1735, 410, 325, 177, 138, 75 and 70, respectively (Fig. 3A). The best selection of NT mutations had been known in D614G positions, adopted via N501Y, P681H, T716I, and A570D within the spike (S) protein (Fig. 3B). Of the known NT mutations, the 4 maximum widespread NT mutations comparable to T1001I, A170BD, I2230T, and T265I had been known within the N gene. Herein this find out about, we noticed 3 maximum NT mutations (e.g., P314L, E1264D, and P218L) within the ORF1b gene. As well as, E, M, ORF3a, ORF6a, ORF7b, ORF8 and ORF9b genes possessed the best selection of NT mutations at P71L, I82T, Q57H, I33T, T4OI, Y73C and Q77E positions, respectively. Different genes with vital NT mutation frequencies at particular websites integrated P71L (E-gene), T4OI (0RF7b), I33T (ORF6a), I82T (M), T40I (ORF7b), Q77E (ORF9b), Q57H (ORF3a) and Y73C (ORF8) (Fig. 3B).Determine 3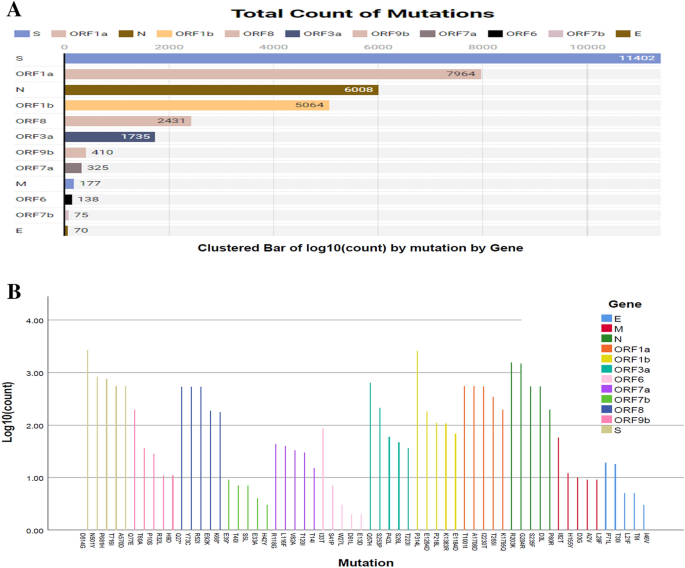 The frequency of nucleotide (NT) mutations discovered all over the SARS-CoV-2 genomes of the deceased COVID-19 sufferers. (A) The selection of conversions respective to express genes or segments of the SARS-CoV-2 genome. (B) The utmost frequency of NT mutations particularly area of the SARS-CoV-2 genome. In each circumstances, particular gene areas had been coloured with frequency.Any other notable discovering of this find out about is the prediction of the NT alterations within the SARS-CoV-2 genomes of quite a lot of countries. We when put next the NT mutational spectra in best 12 international locations (Desk 1), the place the best selection of COVID-19 related deaths had been reported. The frequency of the NT mutations at D614G place within the S gene used to be distinguished in 12 countries with essentially the most vital occurrence of COVID-19 deaths (Desk 1). In a similar fashion, the utmost selection of NT mutations had been known in T1001I, G204R, E1264D, P314L, E92K, Q57H, Q77E, S5L, L29F, S41P, and T40I positions of SARS-CoV-2 genomes. The SARS-CoV-2 genomes sequenced from deceased COVID-19 sufferers of america confirmed most NT mutations at I2230T place in ORF1a, D3L in N, P218L in ORF1b, Y73C in ORF8, Q57H in ORF3a, P10S in ORF9b, A43S in ORF7a, T175M in M, E13D in ORF6, T40I in ORF7b, and P71L in E genes. Against this, genomes sequenced from India confirmed the best NT mutation frequency at D614G in S gene adopted via T1001I in ORF1a, G204R in N gene, K1383R in ORF1b, R52I in ORF8, P42L in ORF3a, Q77E in ORF9b, N38T in ORF7a, L87F in M gene, I14T in ORF6, S5L in ORF7b and T9I in E gene (Desk 1). Those findings indicate that whilst some SARS-CoV-2 NT mutations had been liable for its evolution, a couple of might benefit viral adaptation in a selected demographic distribution. Diversifications in NT mutation patterns in SARS-CoV-2 genomes could also be on account of inhabitants age distribution, gender, host immunity, and socioeconomic stage.Desk 1 The nucleotide (nNT) mutations with the best frequency predicted at quite a lot of loci of SARS-CoV-2 genomes extracted from deceased COVID-19 sufferers of various international locations.Level-specific amino-acid mutations in SARS-CoV-2 genomes of the deceased COVID-19 patientsTo establish deleterious mutations within the SARS-CoV-2 genomes, we analyzed point-specific amino acid (AA) mutations within the genomes of this virus received from deceased COVID-19 sufferers the usage of SIFT, PolyPhen-2, SNAP2, PROVEAN, PredictSNP, and MAPP web-based gear. Deleterious mutations had been significantly analyzed and cross-checked the usage of those gear. A threshold worth of − 2.5 used to be decided to make sure extremely balanced accuracy in defining the deleterious mutation. Due to this fact, mutations having a price smaller than − 2.5 had been known as deleterious33. A few of the AA mutations known, the selection of deleterious and non-deleterious mutations had been 951 and 3199, respectively. The best selection of deleterious AA mutations had been discovered within the ORF1b (n = 338) adopted via ORF1a (n = 236), ORF3a (n = 122). But even so, 49, 45, 42, 40 and 30 deleterious AA mutations had been predicted within the N, ORF8, ORF7a, S and ORF9b segments, respectively. On this find out about, the open studying frames (ORF) of the SARS-CoV-2 genome possessed the next share of deleterious AA mutations than different segments. As as an example, the ORF3a, ORF6, ORF7a, ORF8 and ORF9b harbored > 50.0% deleterious AA mutations. On the other hand, remainder of the segments of the SARS-CoV-2 genomes fewer mutations (< 30) (Desk S1). The whole AA mutations detected within the spike protein of the SARS-CoV-2 genomes of the deceased sufferers are proven in Fig. 4A. On this find out about, the best frequency of AA mutations (31.85%) used to be recorded within the S gene, which is liable for viral pathogenicity. The S gene of the find out about genomes underwent AA mutations at 32 websites (Fig. 4A). Fourteen of those AA mutation websites comparable to V3L, L5Y, L10S, S13L, T19R, P26L, D401, S60A, P82AT, V1201Y204R, S2051, L2231, Y2651 had been predicted in NTD (N-terminal area) fragment, whilst 8 of them (e.g., Q314, G339D, S371F, S373P, F377Y, D405N, K417N, L452R, T478K, and E484Q) had been discovered within the RBD area. The rest 8 websites comparable to A570D, D614G, P681R, N764K, D796Y, N856K, R1000L, and E1188L had been located in numerous spaces of the S protein. The fusion peptide house used to be smartly conserved as a result of no AA mutation hotspots had been came upon (Fig. 4A).Determine 4
The frequency of nucleotide (NT) mutations discovered all over the SARS-CoV-2 genomes of the deceased COVID-19 sufferers. (A) The selection of conversions respective to express genes or segments of the SARS-CoV-2 genome. (B) The utmost frequency of NT mutations particularly area of the SARS-CoV-2 genome. In each circumstances, particular gene areas had been coloured with frequency.Any other notable discovering of this find out about is the prediction of the NT alterations within the SARS-CoV-2 genomes of quite a lot of countries. We when put next the NT mutational spectra in best 12 international locations (Desk 1), the place the best selection of COVID-19 related deaths had been reported. The frequency of the NT mutations at D614G place within the S gene used to be distinguished in 12 countries with essentially the most vital occurrence of COVID-19 deaths (Desk 1). In a similar fashion, the utmost selection of NT mutations had been known in T1001I, G204R, E1264D, P314L, E92K, Q57H, Q77E, S5L, L29F, S41P, and T40I positions of SARS-CoV-2 genomes. The SARS-CoV-2 genomes sequenced from deceased COVID-19 sufferers of america confirmed most NT mutations at I2230T place in ORF1a, D3L in N, P218L in ORF1b, Y73C in ORF8, Q57H in ORF3a, P10S in ORF9b, A43S in ORF7a, T175M in M, E13D in ORF6, T40I in ORF7b, and P71L in E genes. Against this, genomes sequenced from India confirmed the best NT mutation frequency at D614G in S gene adopted via T1001I in ORF1a, G204R in N gene, K1383R in ORF1b, R52I in ORF8, P42L in ORF3a, Q77E in ORF9b, N38T in ORF7a, L87F in M gene, I14T in ORF6, S5L in ORF7b and T9I in E gene (Desk 1). Those findings indicate that whilst some SARS-CoV-2 NT mutations had been liable for its evolution, a couple of might benefit viral adaptation in a selected demographic distribution. Diversifications in NT mutation patterns in SARS-CoV-2 genomes could also be on account of inhabitants age distribution, gender, host immunity, and socioeconomic stage.Desk 1 The nucleotide (nNT) mutations with the best frequency predicted at quite a lot of loci of SARS-CoV-2 genomes extracted from deceased COVID-19 sufferers of various international locations.Level-specific amino-acid mutations in SARS-CoV-2 genomes of the deceased COVID-19 patientsTo establish deleterious mutations within the SARS-CoV-2 genomes, we analyzed point-specific amino acid (AA) mutations within the genomes of this virus received from deceased COVID-19 sufferers the usage of SIFT, PolyPhen-2, SNAP2, PROVEAN, PredictSNP, and MAPP web-based gear. Deleterious mutations had been significantly analyzed and cross-checked the usage of those gear. A threshold worth of − 2.5 used to be decided to make sure extremely balanced accuracy in defining the deleterious mutation. Due to this fact, mutations having a price smaller than − 2.5 had been known as deleterious33. A few of the AA mutations known, the selection of deleterious and non-deleterious mutations had been 951 and 3199, respectively. The best selection of deleterious AA mutations had been discovered within the ORF1b (n = 338) adopted via ORF1a (n = 236), ORF3a (n = 122). But even so, 49, 45, 42, 40 and 30 deleterious AA mutations had been predicted within the N, ORF8, ORF7a, S and ORF9b segments, respectively. On this find out about, the open studying frames (ORF) of the SARS-CoV-2 genome possessed the next share of deleterious AA mutations than different segments. As as an example, the ORF3a, ORF6, ORF7a, ORF8 and ORF9b harbored > 50.0% deleterious AA mutations. On the other hand, remainder of the segments of the SARS-CoV-2 genomes fewer mutations (< 30) (Desk S1). The whole AA mutations detected within the spike protein of the SARS-CoV-2 genomes of the deceased sufferers are proven in Fig. 4A. On this find out about, the best frequency of AA mutations (31.85%) used to be recorded within the S gene, which is liable for viral pathogenicity. The S gene of the find out about genomes underwent AA mutations at 32 websites (Fig. 4A). Fourteen of those AA mutation websites comparable to V3L, L5Y, L10S, S13L, T19R, P26L, D401, S60A, P82AT, V1201Y204R, S2051, L2231, Y2651 had been predicted in NTD (N-terminal area) fragment, whilst 8 of them (e.g., Q314, G339D, S371F, S373P, F377Y, D405N, K417N, L452R, T478K, and E484Q) had been discovered within the RBD area. The rest 8 websites comparable to A570D, D614G, P681R, N764K, D796Y, N856K, R1000L, and E1188L had been located in numerous spaces of the S protein. The fusion peptide house used to be smartly conserved as a result of no AA mutation hotspots had been came upon (Fig. 4A).Determine 4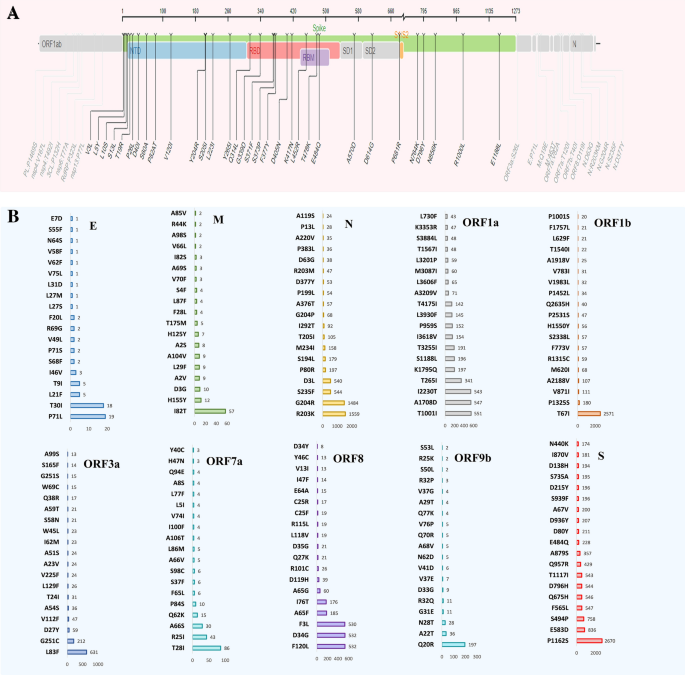 Genomic deletion research in SARS-CoV-2 entire genome sequences of the deceased COVID-19 sufferers. (A) Mapping of amino acid (AA) mutations within the spike (S) glycoprotein of SARS-CoV-2 genome. (B) The AA mutations within the subdomains S1 and S2 (SD1, SD2), N-terminal area (NTD), and receptor binding area (RBD) are highlighted.Excluding for the numerous AA mutation adjustments within the spike protein, there have been notable adjustments within the mutational spectra of alternative proteins as smartly. Compared to the other AA mutational spectra, an enormous selection of repeats had been seen within the ORF1b (T67I, 2571 instances) and N (G204R, 1484; R203K, 1559 instances) segments. There have been extra seven AA mutations discovered to be came about in > 500 sequences comparable to I2230T (543 instances), A1708D (547 instances) and T1001I (551 instances) in ORF1a, L83F (631 instances) in ORF3a, F3L (530 instances), D34G (532 instances) and F120L (532 instances) in ORF8 fragment of the SARS-CoV-2 genome (Fig. 4B).With the march of time, an increasing number of deleterious AA mutations are being detected a few of the genome sequences of SARSCoV-2 particularly the ones sequenced from the deceased COVID-19 sufferers. The highest widespread AA mutations of various proteins happening in numerous continents had been indexed to higher perceive the situation of SARS-CoV-2 mutational tendency relying at the regional issue (Desk 2). The AA mutations happening in additional than two continents are highlighted to concentrate on them. Curiously, D614G mutation in spike protein and S26L mutation in OF3a protein had been discovered to happen within the SARS-CoV-2 genomes in all continents. Any other noteworthy AA mutation, A1918V, came about in each Asia and North American areas while a somewhat other mutation, A1818L discovered within the African area. On the other hand, the ORF8 and ORF9b fragments confirmed no similarity of mutational alignment around the regional barrier (Desk 2).Desk 2 Maximum widespread amino acid (AA) mutations predicted at quite a lot of loci of SARS-CoV-2 genome received from deceased COVID-19 sufferers of the 5 continents.Results of mutations on protein functionsFinally, we thought to be the deleterious signature mutations to guage the adjustments in proteins as organic purposes the usage of PROVEAN, PolyPhen-2, and Are expecting SNP gear (Fig. S1). We discovered the best PROVEAN ranking of − 13.22 in case of W45S and W45R deleterious mutations, and a minimal − 12.278 for the W45L mutation of the ORF8 gene (Fig. 5A). Curiously, those 3 destructive mutations had been known in the similar ORF8 area of SARS-CoV-2 genomes the usage of different gear. The use of those 3 gear, G18V, W45S, I33T, P30L, and Q418H had been known because the widespread mutations that are liable for defining every clade as all of them are deleterious and risky. The use of Are expecting SNP, we concurrently predicted the best selection of destructive mutations (n = 1875) at Q57H within the ORF3a gene (Fig. 5B). During the PolyPhen-2, we detected the best selection of deleterious mutations at D160Y (n = 1559) within the M gene and G204R (n = 1448) and D3L (n = 540) within the N gene. A majority of these deleterious mutations had a PolyPhen-2 ranking of one, while the sensitivity and specificity had been 0 and 1, respectively. Those findings point out that variations in mutations in distinct areas will most probably have an effect on protein serve as. Best mutations towards the Are expecting SNP ranking are visualized in Fig. 5C. The mutations came about within the ORF8 phase comparable to W45L andW45S scored essentially the most unfavorable values in keeping with Are expecting SNP prediction fashion the place each scored lower than -12. No different mutation of this phase or different proteins had scored such unfavorable ratings all over the mutational spectra (Fig. 5C).Determine 5
Genomic deletion research in SARS-CoV-2 entire genome sequences of the deceased COVID-19 sufferers. (A) Mapping of amino acid (AA) mutations within the spike (S) glycoprotein of SARS-CoV-2 genome. (B) The AA mutations within the subdomains S1 and S2 (SD1, SD2), N-terminal area (NTD), and receptor binding area (RBD) are highlighted.Excluding for the numerous AA mutation adjustments within the spike protein, there have been notable adjustments within the mutational spectra of alternative proteins as smartly. Compared to the other AA mutational spectra, an enormous selection of repeats had been seen within the ORF1b (T67I, 2571 instances) and N (G204R, 1484; R203K, 1559 instances) segments. There have been extra seven AA mutations discovered to be came about in > 500 sequences comparable to I2230T (543 instances), A1708D (547 instances) and T1001I (551 instances) in ORF1a, L83F (631 instances) in ORF3a, F3L (530 instances), D34G (532 instances) and F120L (532 instances) in ORF8 fragment of the SARS-CoV-2 genome (Fig. 4B).With the march of time, an increasing number of deleterious AA mutations are being detected a few of the genome sequences of SARSCoV-2 particularly the ones sequenced from the deceased COVID-19 sufferers. The highest widespread AA mutations of various proteins happening in numerous continents had been indexed to higher perceive the situation of SARS-CoV-2 mutational tendency relying at the regional issue (Desk 2). The AA mutations happening in additional than two continents are highlighted to concentrate on them. Curiously, D614G mutation in spike protein and S26L mutation in OF3a protein had been discovered to happen within the SARS-CoV-2 genomes in all continents. Any other noteworthy AA mutation, A1918V, came about in each Asia and North American areas while a somewhat other mutation, A1818L discovered within the African area. On the other hand, the ORF8 and ORF9b fragments confirmed no similarity of mutational alignment around the regional barrier (Desk 2).Desk 2 Maximum widespread amino acid (AA) mutations predicted at quite a lot of loci of SARS-CoV-2 genome received from deceased COVID-19 sufferers of the 5 continents.Results of mutations on protein functionsFinally, we thought to be the deleterious signature mutations to guage the adjustments in proteins as organic purposes the usage of PROVEAN, PolyPhen-2, and Are expecting SNP gear (Fig. S1). We discovered the best PROVEAN ranking of − 13.22 in case of W45S and W45R deleterious mutations, and a minimal − 12.278 for the W45L mutation of the ORF8 gene (Fig. 5A). Curiously, those 3 destructive mutations had been known in the similar ORF8 area of SARS-CoV-2 genomes the usage of different gear. The use of those 3 gear, G18V, W45S, I33T, P30L, and Q418H had been known because the widespread mutations that are liable for defining every clade as all of them are deleterious and risky. The use of Are expecting SNP, we concurrently predicted the best selection of destructive mutations (n = 1875) at Q57H within the ORF3a gene (Fig. 5B). During the PolyPhen-2, we detected the best selection of deleterious mutations at D160Y (n = 1559) within the M gene and G204R (n = 1448) and D3L (n = 540) within the N gene. A majority of these deleterious mutations had a PolyPhen-2 ranking of one, while the sensitivity and specificity had been 0 and 1, respectively. Those findings point out that variations in mutations in distinct areas will most probably have an effect on protein serve as. Best mutations towards the Are expecting SNP ranking are visualized in Fig. 5C. The mutations came about within the ORF8 phase comparable to W45L andW45S scored essentially the most unfavorable values in keeping with Are expecting SNP prediction fashion the place each scored lower than -12. No different mutation of this phase or different proteins had scored such unfavorable ratings all over the mutational spectra (Fig. 5C).Determine 5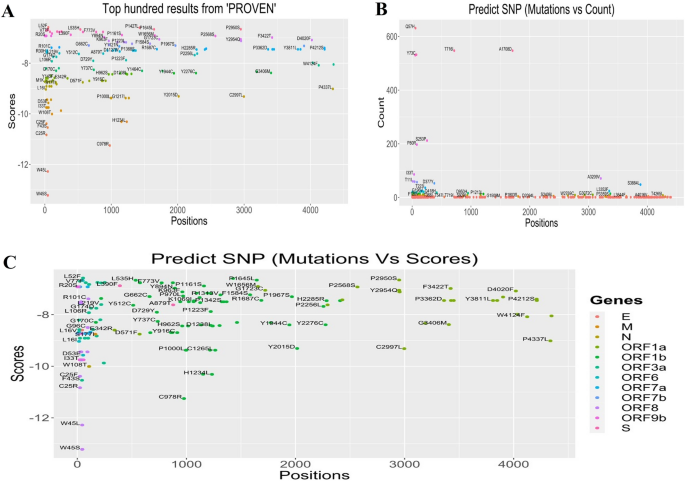 Rankings of various mutations all over the SARS-CoV-2 genomes sequenced from deceased COVID=19 sufferers. (A) Best hundred mutations predicted via PROVEAN software. (B) Overall frequency of the highest mutations predicted via Are expecting SNP software. (C) Prediction of deleterious mutations via Are expecting SNP.
Rankings of various mutations all over the SARS-CoV-2 genomes sequenced from deceased COVID=19 sufferers. (A) Best hundred mutations predicted via PROVEAN software. (B) Overall frequency of the highest mutations predicted via Are expecting SNP software. (C) Prediction of deleterious mutations via Are expecting SNP.

, Micron (MU) Achieve After Foxconn Reviews Robust Effects")










